Paciente de sexo masculino, de 63 años de edad, que consulta por cuadro de diarrea crónica, hiporexia no selectiva y pérdida de peso de 13 Kg en 5-6 meses de evolución. Durante las últimas 48 hs refiere haber presentado…
Paciente de sexo masculino, de 63 años de edad, que consulta por cuadro de diarrea crónica, hiporexia no selectiva y pérdida de peso de 13 Kg en 5-6 meses de evolución. Durante las últimas 48 hs refiere haber presentado 2 episodios de melena, por lo que se decide a consultar.
Antecedentes personales: Ex tabaquista. Poliartralgias de 3 años de evolución en tratamiento con glucosamina y AINES. Dos episodios de afasia en el último año, interpretado como de origen vascular y medicado con AAS. Episodio de desprendimiento retiniano hace un año. Estado depresivo y cambios notorios de la personalidad durante el último año.
Examen físico: Aspecto adelgazado, impresiona enfermo. Palidez cutáneo mucosa. Normotenso, afebril. Abdomen plano excavado, blando, depresible e indoloro sin visceromegalias. No se palpan adenopatías.
Laboratorio de ingreso: Hto: 28%; Gl. Blancos: 6550/mm3; Plaquetas: 505.000/mm3; Glicemia: 100 mg%; Urea: 27 mg%; Creatinina: 0,65 mg%; Bilirubina Total: 0,47 mg% (Directa 0,1); TGO 12 UI/L; TGP: 7 UI/L; FAL: 296 mg%; GGT: 25 mg%; Tp 15,5 seg; kPTT 25 seg; Albúmina: 2,7 g%; LDH: 216 UI/L.
Ecografía abdominal: Imagen quística de 20 mm, de aspecto simple, en parénquima hepático como único hallazgo anormal.
Tomografía computada de abdomen y pelvis con contraste: Estructuras quísticas hepáticas. Quiste seroso simple renal izquierdo. Múltiples estructuras ganglionares mesentéricas y retroperitoneales predominantemente en raíz del mesenterio, algunas de ellas de rango megálico. Escasa cantidad de líquido libre en FSD.
Se realizan estudios endoscópicos:
VEDA: Lesión compatible con angiodisplasia aislada en antro. En bulbo, y hasta 3era porción duodenal, múltiples lesiones ulceradas pequeñas de 2-3 mm con base limpia y edema.
Videocolonoscopía: Se progresa hasta ciego, observándose mucosa normal, con resto de materia fecal liquida, con aspecto de melena. Hemorroides internas.
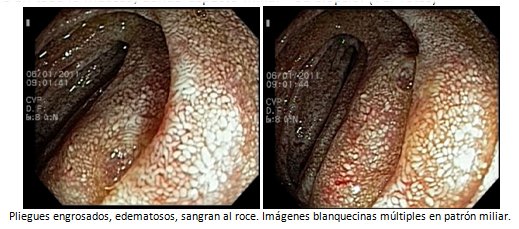
Endoscopía de Intestino Delgado con colonoscopio: Se progresa 70 cm desde píloro (yeyuno proximal-medio) ailsdas zonas de mucosa denudada, friabilidad al roce, y lesiones blanco-grisáceas distribuidas en toda la mucosa, dando aspecto miliar – Se biopsia. (Ver fotos)
Debido a los resultados se infiere el diagnóstico posible de Enfermedad de Whipple (EW), es evaluado en conjunto con servicio de Neurología que realiza punción lumbar y demuestra un líquido cefalorraquídeo de características normales.
Se envían muestras de Sangre, Líquido Cefalorraquídeo, saliva y tejido de Intestino delgado para determinación de PCR Thropheryma whippleii (Tw) que resultaron negativas.
Se interpreta finalmente como EW posible.
Se comienza tratamiento antibiótico con Ceftriaxona (CTX) IV por 14 días y se continúa con Trimetoprima- Sulfametoxazol (TMS) 160/800 mg cada 12 hs por un período de 1 año.
El paciente evolucionó favorablemente, con respuesta clínica, endoscópica e histológica satisfactoria. Se suspendió tratamiento antibiótico, y por el momento no ha presentado recaídas en los síntomas.
Enfermedad de Whipple
Introducción
Es un trastorno multisistémico, antiguamente letal, caracterizado principalmente por diarrea malabsortiva, poliartralgias, síndrome febril prolongado y pérdida significativa y sostenida de peso. Para que se produzca se considera que son necesarios 2 elementos, la infección por el Thropheryma whippleii (Tw) y el padecimiento de algún tipo de defecto inmunológico no del todo aclarado, subclínico, que predispone a padecerla. Debido a que es extremadamente infrecuente, y a que sus síntomas pueden ser muy variados, se cree que existe un importante subdiagnóstico.
Epidemiología
Hasta el año 2009 no existían más de 1500 casos reportados en la literatura mundial, la mayoría de los cuales fueron expuestos en los últimos 20 años debido principalmente a la evolución en la metodología diagnóstica. Afecta más frecuentemente a varones caucásicos de unos 50 años en promedio. Ciertos factores epidemiológicos, como la adquisición en áreas rurales por personas que trabajan en la remoción de tierras, agricultores, etc. fueron formulados pero no demostrados.
Microbiología
El Tw es un microorganismo incluido en el género de Actinomicetales, caracterizados por su hábitat en tierra, aguas servidas y también en animales sanos (incluido el hombre). Se duplica de manera muy lenta, y obtiene condiciones óptimas de replicación en medios ácidos como los lisosomas macrofágicos, lo que constituye uno de los mecanismos de evasión inmunológica. El cultivo in vitro es extremadamente dificultoso y fue logrado por Didier Raoult casi 100 años después de la primer descripción de la enfermedad a cargo de George Whipple en 1907. La adquisición fue ligada por mucho tiempo a la exposición ambiental, pero trabajos realizados en áreas de alta endemicidad como el África subsahariana sugieren un posible mecanismo fecal-oral directo para la misma.
Patogenia
Como fuera mencionado anteriormente, es necesario factores del huésped para que la enfermedad se desarrolle, además de la infección. Dicha predisposición ha sido expuesta de manera experimental, estudiando el fenotipo de las células inmunológicas in vitro. Se ha evidenciado una transformación inmunotolerante de las células macrofágicas (fenotipo M2) y dendríticas, impulsado por un medio rico en linfocinas y citoquinas que inducen dicha transformación (IL-10, IL-4; CCL-18; IL-16).
Un grupo de investigadores argentinos del Hospital Bonorino Udaondo desestimó, mediante la evaluación de 14 pacientes con EW vs 174 controles sanos, la asociación con el HLA-B27.
Ciertos aspectos no han sido aun dilucidados, principalmente la vía de acceso de los micro-organismos al córion, los factores que inducen una primer respuesta inmunotolerante por parte de los macrófagos y la vía por la cual se infectan los monocitos reclutados (ver figura).

Manifestaciones Clínicas
La primer descripción de la enfermedad en 1907 destacaba la presencia de diarrea crónica malabsortiva, pérdida de peso, debilidad y poliartralgias, que culminó con el fallecimiento del paciente al cabo de 5 años. Exceptuando lo del desenlace fatal, ese mismo cuadro clínico es la forma de presentación más frecuente, en un 80-90% de los casos aproximadamente.
El compromiso articular, más frecuentemente confundido con cuadros como Artritis Reumatoidea o Artrosis, afecta tanto a medianas como grandes articulaciones, muchas veces sin un marcado componente inflamatorio.
En ocasiones pueden evidenciarse adenomegalias periféricas, pero más frecuentemente se las halla mediante estudios por imágenes en proyección de vasos mesentéricos y retroperitoneales (50%).
La fiebre prolongada es un signo frecuente, pero no excluyente (38%).
Otras manifestaciones importantes, pero menos frecuentes e inespecíficas, son las oculares (uveítis anterior y posterior), la melanodermia, el derrame pleural y el compromiso endocardico.
La presentación con melena ha sido descripta de manera aislada en un reporte de caso.
Párrafo aparte merecen las frecuentes manifestaciones neurológicas, las cuales condicionan una peor respuesta al tratamiento y por ende mayor morbimortalidad. Estas pueden ser muy variadas, pero las más frecuentes son las que afectan a la esfera cognitiva y emocional como depresión, disminución del rendimiento intelectual, alteraciones de la personalidad y hasta cuadros de demencia. En ocasiones se pueden presentar trastornos del movimiento como ataxia cerebelosa, mioclonías y convulsiones. Una manifestación muy poco frecuente, pero patognomónica, es la combinación de un cuadro de miorritmia oculomasticatoria con oftalmoplejía supranuclear. Cabe aclarar que, el compromiso del Sistema Nervioso Central por el Tw, puede ser en el contexto de una enfermedad clásica, como de manera aislada. Las imágenes por Resonancia Magnética suelen mostrar áreas de inflamación en múltiples localizaciones, y en ocasiones lesiones ocupantes de espacio.
Para finalizar, es muy poco frecuente, pero posible, la presencia de compromiso intestinal aislado (< 15%), por lo que la sola presencia de diarrea malabsortiva debe hacer reflexionar sobre algún diagnóstico alternativo.
Diagnóstico
Arribar al diagnóstico de esta entidad es un verdadero desafío, debido principalmente a la inespecificidad de las manifestaciones clínicas, bioquímicas e imagenológicas. Habitualmente es tomada en consideración luego de que otras entidades han sido debidamente descartadas (Celiaquía, hipertiroidismo, enfermedad inflamatoria, VIH/SIDA, etc.).
La endoscopía digestiva es una herramienta crucial debido a que permite observar las alteraciones macroscópicas de la mucosa (pliegues engrosados, hiperémicos, que sangran fácilmente al roce, con un patrón miliar sobre la superficie representando a la intensa linfangiectasia que le dio el primer nombre de lipodistrofia intestinal). Además, es la técnica que habilita la toma de biopsias múltiples que son las que demostrarán los signos típicos en la mayoría de los casos (edema del córion, infiltrado mononuclear con numerosos macrófagos cargados de material PAS + resistente a la disolución con diastasa y Ziehl Neelsen negativo). La toma de biopsias de otros tejidos como ganglios linfáticos, hígado y médula ósea, en el contexto de un síndrome febril prolongado, pueden mostrar granulomas no caseificantes mal conformados y la mayoría de las veces PAS -, lo cual no debe excluir el diagnóstico.
Para afirmar o descartar EW debe realizarse inmunohistoquímica con anticuerpos de conejo dirigidos contra Tw (disponible solo en centros experimentales). Otra técnica directa de diagnóstico, de mayor aplicabilidad clínica, es la Reacción en Cadena de la Polimerasa en tiempo real (RT-PCR), la cual puede ser utilizada en tejidos fijados y fluidos corporales como sangre, saliva y líquido cefalorraquídeo. Su positividad indica con gran seguridad la presencia del bacilo, pero no determina la existencia de la enfermedad en ausencia del contexto sintomatológico adecuado.
Para resumir, la situación que se presenta con mayor frecuencia es la que se muestra en el siguiente gráfico.

Tratamiento
Es sin duda el área con mayor número de interrogantes. Desde 1950, 2 años después de que se descubra que la enfermedad tenía una base infectológica, la EW se trata con antibióticos. El primer intento fue con cloranfenicol. Inmediatamente, y por muchos años más, le siguió la combinación de Penicilina con Estreptomicina y las tetraciclinas por periodos prolongados (1 a 2 años) con grados variables de respuesta y recaídas (estas últimas en alrededor del 30%).
En la actualidad el tratamiento más aceptado, a pesar de algunos disensos, es la combinación de CTX 2 gr/día EV por 14 días con TMS 160/800 mg cada 12 hs VO por 1 a 2 años. Dicho esquema se justifica el rápido comienzo de acción con la CTX y la facilidad del traspaso de la barrera hematoencefálica por ambos compuestos, lo cual reduciría el número de recaídas por compromiso del SNC. El único trabajo clínico randomizado sobre 40 pacientes comparó CTX/TMS vs Meropenem/TMS, no demostrando diferencias significativas en los objetivos evaluados.
Raoult y colaboradores cuestionan dichos resultados presentando el fracaso del tratamiento con TMS en 14 pacientes en un seguimiento por 3 años. Atribuyen dichos resultados negativos a que, en primer lugar el Tw es resistente natural a la Trimetoprima, en segundo lugar a que se han detectado mutaciones que lo hacen resistente al Sulfametoxazol; a que, a pesar de lo expresado por otros autores, existen casos de recaídas y, por último, a que se han suspendido tratamientos por efectos adversos y reconstitución inmune.
Por todo lo antedicho, y por investigaciones realizadas sobre susceptibilidad in vitro, existe una tendencia a modificar el esquema de tratamiento a la utilización combinada de Doxiciclina con Hidroxicloroquina más el agregado de Sulfadiazina en quienes presenten compromiso neurológico.
Conclusión
El paciente presentado en esta ocasión constituye un caso con predominio franco de manifestaciones típicas de EW, pero con el agregado de otras de muy infrecuente aparición, como melena. Queda sin definir si algunas de las manifestaciones restantes, como el desprendimiento retiniano y los episodios de afasia, son ocasionadas por la misma enfermedad o constituyen manifestaciones de otra entidad no diagnosticada.
Debido a su infrecuencia, y a la elevada morbimortalidad que conlleva el subdiagnóstico, es necesario un alto nivel de sospecha ante cuadros clínicos similares a los descriptos, luego de lo cual el diagnóstico suele ser más sencillo de lo previsto.
Sin dudas que la gran materia pendiente en EW es la obtención de mayor cantidad y calidad de evidencia respecto al tratamiento, el cual debería lograr, en condiciones óptimas, tasas de curación muy cercanas al 100%, y con un mínimo número de recaídas.
Bibliografía:
- Mandell, Douglas y Bennett. Enfermedades Infecciosas, principios y práctica. Cap97. Pags 1306-1310.
- Fenollar F; Puéchal X and Raoult D. Whipple´s Disease. N Eng J Med 2007;365:55-66.
- Dutly F and Altwegg M. Whipple’s Disease and “Tropheryma whippelii”. Clin Microbiol Rev 2001;14:561-83.
- Pyrgioti M and Kyriakidis A. Whipple´s disease: a review. Ann Gastroenterol 2004;17:43-50.
- Bermejo P and Burgos A. Enfermedad de Whipple y sistema nervioso central. Med Clin (Barc) 2006;127:379-85.
- Schij LJ; Becx MCJM; De Bruin PC and Van der Vegt SGL. Whipple’s disease: easily diagnosed, if considered. Neth J Med 2008;66:392-96.
- Panegyres PK; Edis R; Beaman M and Fallon M. Primary Whipple’s disease of the brain: characterization of the clinical syndrome and molecular diagnosis. Q J Med 2006;99:609-623.
- García Bernárdez AM; García Díez AI; Álvarez Cuesta CC and cols. Enfermedad de Whipple. Dos nuevos casos de una enfermedad infradiagnosticada. An Med Interna (Madrid) 2005;22:231-234.
- Hanneke K. A. Knaapen and Pilar Barrera. Therapy for Whipple’s disease. J Antimicrob Chemother 2007;60:457-8.
- Desnues B; Ihrig M; Raoult D and Mege JL. Whipple’s Disease: a Macrophage Disease. Clin Vaccine Inmunol 2006;13:170-78
- Freeman HJ. Tropheryma whipplei infection. World J Gastroenterol 2009; 15(17):2078-2080
- Raoult D et al. Resistance to trimethoprim/sulfamethoxazole and Tropheryma whipplei.Int J Antimicrob Agents 2009;34:255-9.
- Raoult D et al. Failure and relapse after treatment with Trimethoprim/Sulfamethoxazole in classic Whipple Disease. J Antimicrob Chemother 2010;65:2005-12
- Feurle GE, Junga NS, Marth T. Efficacy of ceftriaxone or meropenem as initial therapies in Whipple´s disease. Gastroenterology 2010;138:478-86
- Bai JC, Mota AH, Mauriño E, et al. Class I and class II HLA antigens in a homogeneous Argentinian population with Whipple’s disease: lack of association with HLA-B 27. Am J Gastroenterol. 1991;86(8):992.